
Синдром Алажиля
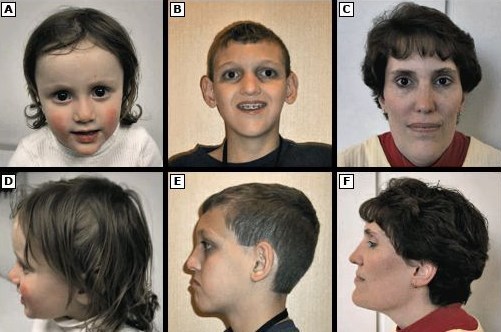
Синдром Алажиля
Другие названия болезни
- Синдром Алажиля-Ватсона
- артериально-печеночная дисплазия
- холестаз с периферическим легочным стенозом
- синдромальная недостаточность желчных протоков
Общий диагноз
Синдром Алажиля представляет собой аутосомно-доминантное заболевание, характеризующееся холестазом, пороками сердца, аномалиями скелета, аномалиями глаз и характерным фенотипом лица, обычно с 5 основными клиническими аномалиями. Холестаз является прямым следствием определенных нарушений в желчных протоках.
Около 39% пациентов имеют заболевания почек, особенно почечную дисплазию. Дополнительные симптомы включают шумы в сердце, врожденные пороки сердца, различия в позвонках (позвоночнике), утолщение кольца, которое в норме выравнивает роговицу (задний эмбриотоксон) в глазу, и характерные черты лица.
У большинства людей с синдромом Алажиля есть мутации в одной копии гена JAG1. Небольшой процент (менее 1 процента) пациентов имеют мутации в гене NOTCH2. Эти мутации наследуются как аутосомно-доминантные признаки, однако примерно в половине случаев мутация возникает как новое изменение («de novo») у индивидуума и не наследуется от родителя. Текущая расчетная заболеваемость ALGS составляет от 1:30 000 до 1:45 000 без различий по полу.
Симптомы болезни
Симптомы и тяжесть синдрома Алажиля могут сильно различаться у разных людей, даже у членов одной семьи. У некоторых людей может быть легкое, едва заметное расстройство; это может быть серьезная форма заболевания, которая может вызвать потенциально опасные для жизни осложнения у других людей. Важно отметить, что у пострадавших людей могут отсутствовать все симптомы, описанные ниже. Затронутые люди должны поговорить со своими врачами и медицинскими бригадами о своем конкретном случае, связанных симптомах и общем прогнозе. Синдром Алажиля может быть связан с аномалиями в печени, сердце, глазах, скелете, почках и других системах органов организма.
Основным проявлением синдрома Алажиля является поражение печени, которое чаще возникает в первые три месяца жизни. Однако у лиц с легким поражением печени, может быть не диагностировано в более позднем возрасте.
Поражение печени при синдроме Алажиля может варьироваться от желтухи или легкого холестаза, если он присутствует, до тяжелого, потенциально прогрессирующего заболевания печени, которое может вызвать печеночную недостаточность.
Из-за уменьшенного количества желчных протоков у людей с синдромом Алажиля могут развиться желтуха и холестаз, как правило, в течение первых четырех месяцев жизни. Холестаз относится к уменьшению или ингибированию оттока желчи из печени. Холестаз может вызывать пожелтение кожи и белков глаз (желтуха), плотный, бледный стул непосредственно под поверхностью кожи, темную мочу, жирные язвы или опухоли (ксантомы), аномальное увеличение печени (гепатомегалию) и/ или увеличение селезенки (спленомегалия). Жиры и жирорастворимые витамины (А, D, У больных детей также может наблюдаться отставание в росте и плохое развитие, поскольку они не могут должным образом усваивать витамины Е и К. Нарушение всасывания жизненно важных питательных веществ также может привести к рахиту, размягчению и ослаблению костей (дефицит витамина D), проблемам со зрением (дефицит витамина А), плохой координации и задержке развития (дефицит витамина Е) и проблемам со свертываемостью крови (дефицит витамина К).
Генетические факторы
Синдром Алажиля вызывается мутациями в одном из двух генов: в гене JAG1 или в гене NOTCH2.
Мутации гена JAG1 выявлены более чем в 88% случаев. Мутации в гене NOTCH2 составляют менее 1% случаев. Эти мутации наследуются как аутосомно-доминантные признаки. В некоторых случаях мутации происходят случайно из-за спонтанного генетического изменения (т.е. новой мутации).
Исследователи определили, что большинство случаев синдрома Алажиля возникает из-за мутаций гена JAG1, расположенного на коротком плече (9) хромосомы 20 (20p12).
Примерно в 6-7 процентах случаев синдрома Алажиля у людей наблюдается полная делеция или потеря гена JAG1. У этих людей может быть более тяжелая форма синдрома Алажиля, в зависимости от того, насколько велика делеция и какие другие гены на хромосоме 20. Исследователи определили, что ген NOTCH2 расположен на коротком плече хромосомы 1 (1p13-p11).
Заболеваемость
Синдром Алажиля поражает мужчин и женщин в равной степени. Заболеваемость синдромом Алажиля оценивается примерно в 1 случай на 30 000–45 000 человек в общей популяции. Некоторые случаи синдрома Алажиля могут быть не диагностированы или диагностированы неправильно, что затрудняет определение истинной частоты синдрома Алажиля среди населения в целом.
Стандартные методы лечения болезни
Лечение синдрома Алажиля направлено на конкретные симптомы, которые проявляются у каждого человека.
Лечение может потребовать скоординированных усилий группы специалистов. Педиатрам, гастроэнтерологам, кардиологам, офтальмологам и другим медицинским работникам может потребоваться систематическое и всестороннее планирование лечения ребенка.
Лица с синдромом Алажиля должны пройти базовую эхокардиограмму (УЗИ сердца) для УЗИ брюшной полости и сканирующее исследование глаз (офтальмологическое) для выявления аномалий сердца, брюшной полости и почек. Сканирование кровеносных сосудов головы (МРТ/МРА) рекомендуется для детей, достаточно взрослых, чтобы иметь возможность высидеть исследование без необходимости специфических симптомов, анестезии или седации. Дополнительная терапия витаминами и питательными веществами необходима для людей с нарушением всасывания. Это лечение A, D, В него могут входить восстанавливающие витамины Е и К.
Маленьким детям можно давать смесь со среднецепочечными триглицеридами, потому что эта форма жира лучше усваивается людьми с синдромом Алажиля и холестазом. В некоторых случаях больным детям может потребоваться введение дополнительных калорий через трубку, идущую из носа в желудок (назогастральный зонд), или через трубку, вводимую непосредственно в желудок через небольшой разрез в брюшной стенке и желудке (гастростомическая трубка).
Конкретные методы лечения синдрома Алажиля могут включать:
- Лекарство, усиливающее отток желчи из печени
- Лекарство для облегчения ощущения зуда
- Уход за кожей, например, увлажняющие средства для уменьшения зуда.
- витаминные добавки
- высококалорийные пищевые добавки
- Операция по перенаправлению желчи, чтобы сохранить меньше билирубина в крови
- Трансплантация печени при печеночной недостаточности
Питание, диета и питание при синдроме Алажиля
Как синдром Алажиля влияет на питание?
Снижение поступления желчи в тонкий кишечник может вызвать проблемы с перевариванием жиров и всасыванием жирорастворимых витаминов , таких как витамины A, D, E и K. Эти проблемы могут вызывать недоедание и играть роль в вызывая осложнения , такие как проблемы с костями, проблемы роста, задержка полового созревания или неспособность развиваться.
Что следует есть людям с синдромом Алажиля?
Получение достаточного количества питательных веществ важно для людей с синдромом Алажиля, особенно для младенцев и детей. Получение достаточного количества питательных веществ может помочь ребенку с синдромом Алажиля максимально вырасти. Если у вас или вашего ребенка синдром Алажиля, поговорите с врачом или диетологом о плане здорового питания.
Врачи и диетологи могут порекомендовать
высококалорийная диета, содержащая углеводы и жиры, называемые триглицеридами со средней длиной цепи (ТСЦ), которые легче усваиваются людьми с синдромом Алажиля.
специальная смесь для младенцев, содержащая МСТ
регулярные проверки веса и роста, измерения роста и развития в детстве
регулярные анализы крови для проверки уровня витаминов и отдельные добавки жирорастворимых витаминов A, D, E и K и других питательных веществ по мере необходимости
В некоторых случаях дети с синдромом Алажиля не могут съесть достаточно пищи, чтобы получить энергию, необходимую для роста. Врач может порекомендовать использовать зонд для кормления, чтобы доставлять питательные вещества непосредственно в желудок ребенка. Питательный зонд может представлять собой назогастральный зонд, который вводится через нос, или гастростомический зонд, который вводится через небольшой хирургический разрез в брюшной полости.
Источники:
Этот контент предоставляется как услуга Национального института диабета, болезней органов пищеварения и почек (NIDDK), входящего в состав Национального института здоровья
Медицина Хопкинса, синдром Алажиля, 2022 г.